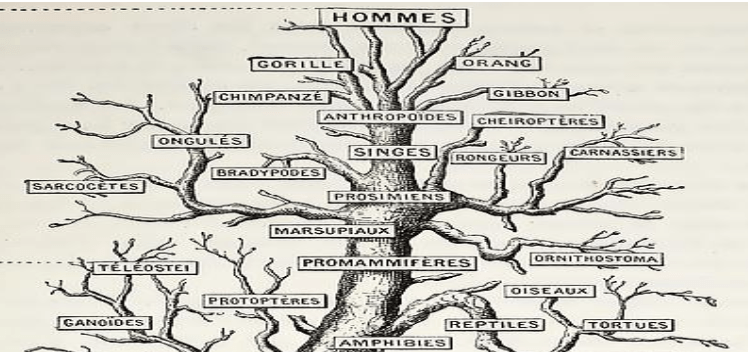
Most people have heard the expression “the tree of life”. Depending on your background, it may give reference to a tree that offers immortality to those who eat its fruit, or be the spring from which new life emerges. In evolutionary biology, the tree of life is an analogy to describe “when” different organisms emerged over the course of our planets history and “how” those organisms are related to one another. In this analogy, the leaves represent the various organisms that have existed through time, the branches represent the relationships between those organisms, and the root of the tree represents the origins of life itself. Occasionally, shorter branches that don’t reach the trees crest represent species or groups long extinct. The organization of leaves, branches, and root form a subdivision of science within evolutionary biology called “systematics”.


In most cases, systematists are not interested in looking at the entire “tree of life” but just a snapshot of a particular group of organisms. To do this, imagine breaking a primary branch from this tree of life; which with it brings its own set of branches and leaves. If we bend the branches into perfect right angles, we would get a structure called a cladogram or phylogenetic tree (Fig. 1). In both, our organisms (i.e. leaves) would be called nodes. These nodes would be connected by branches which represent the relatedness among groups. In addition, branches may represent evolutionary time; illustrating the difference between trees and cladograms. In phylogenetic trees, the length represents time (see the “modern” tree); this differs from cladograms where branches lengths are arbitrary. When a single bifurcated branch connects two nodes together, we would call those nodes “sister” to one another. If we move down the branches connecting these two sister nodes, we reach a new structure referred to as the internal node. Internal nodes are found right where one branch bifurcates into two, and we refer to this internal node as the common ancestor of the nodes at the branch tips. For example, in our Figure 2 tree, you can imagine the internal node linking apes and rodents to be some small, 4-legged hairy creature that existed in ancient times. Some of its descendants continued on to become modern rodents, while others evolved the ability to stand partially upright and led to the gorillas and chimpanzees we know so well. But how do we build these cladograms and what evidence can we use to show what we’ve built is representative of what’s actually occurred through time?

In our rodent/ape example, we can infer the common ancestor was a 4 legged, warm-blooded, hairy organism because those are character traits shared by both apes and rodents. Character traits in general are great at helping us infer the relationships within a cladogram. These traits are usually morphological/physiological in nature, and include things like the presence of a spine, having a prehensile tail, or being bilaterally symmetrical. Using character traits for cladogram reconstruction is especially useful for fossils, where genetic material may not be available for analysis. Nowadays, genetic material can be used for more robust cladogram reconstruction by comparing similarities between the DNA of organisms. However, before the advent DNA analysis, the use of character traits was a common and widely accepted tool for creating cladograms.
 Let’s take Figure 2, for example. Among the organisms listed, the presence of spine is a trait found in all groups but the sea squirt, so we can place it at the base of our tree. We then place a mark above its common ancestor to denote where we believe the presence of a spine emerged. If we continue in this fashion, we could mark up a possible cladogram as such: A mark for the emergence of a jaw above the common ancestor of lampreys; a mark for appearance of 4 limbs above the common ancestor of angelfish; a mark for the emergence of fur above the common ancestor of frogs. What these character trait marks tell us, is that while the character may not have existed in the common ancestor of the node of interest (as one of the nodes does not contain said character) the character likely came into existence somewhere before the next common ancestor in the tree appears. We can count a total of four character steps we used to construct this tree; and the process of counting character steps segways into the topic of parsimony.
Let’s take Figure 2, for example. Among the organisms listed, the presence of spine is a trait found in all groups but the sea squirt, so we can place it at the base of our tree. We then place a mark above its common ancestor to denote where we believe the presence of a spine emerged. If we continue in this fashion, we could mark up a possible cladogram as such: A mark for the emergence of a jaw above the common ancestor of lampreys; a mark for appearance of 4 limbs above the common ancestor of angelfish; a mark for the emergence of fur above the common ancestor of frogs. What these character trait marks tell us, is that while the character may not have existed in the common ancestor of the node of interest (as one of the nodes does not contain said character) the character likely came into existence somewhere before the next common ancestor in the tree appears. We can count a total of four character steps we used to construct this tree; and the process of counting character steps segways into the topic of parsimony.
 Parsimony suggests that the most likely tree is the one which requires the least number of steps to construct. To illustrate this point, let’s see what happens when we switch the lamprey and angelfish on our cladogram (Fig. 3). In this alternate cladogram we have five steps, since the lamprey had to independently lose the appearance of jaws. Systematists who follow the rules of parsimony would say this tree is less likely, as it requires are greater number of evolutionary steps to occur.
Parsimony suggests that the most likely tree is the one which requires the least number of steps to construct. To illustrate this point, let’s see what happens when we switch the lamprey and angelfish on our cladogram (Fig. 3). In this alternate cladogram we have five steps, since the lamprey had to independently lose the appearance of jaws. Systematists who follow the rules of parsimony would say this tree is less likely, as it requires are greater number of evolutionary steps to occur.
The caveat to parsimony is while trees with the least number of steps are accepted, this doesn’t necessarily mean the cladogram is a correct representation of what occurred through evolutionary time. It is possible that the “true” tree had more character appearances/losses than would be expected out of parsimony. In addition, the number of possible trees you can create grows exponentially with the number of nodes on your cladogram; with multiple trees having an equal number of steps. That’s why it’s important to compare with genetic information if possible or look towards other characters to see if the data is robust across multiple comparisons. The more congruent different character states or genetic data are, the more likely that the tree you are looking at is the one most representative of what occurred through evolutionary time.
